
David Hoksza zkoumá proteiny, není ovšem biolog, nýbrž informatik. Na katedře softwarového inženýrství vyvíjí nástroje, které umožňují vizualizovat proteinové struktury nebo na nich identifikovat klíčová místa, kde se vážou léčiva. Zatímco software z dílny jeho výzkumné skupiny využívají vědci po celém světě včetně velkých evropských molekulárních databází, na Univerzitě Karlově jeho know-how oceňují hlavně studenti. Díky Hokszovi a jeho kolegům se totiž na Matfyzu a Přírodovědecké fakultě již několik let vyučuje bioinformatika, moderní multidisciplinární obor, který kombinuje biologii s informatikou a matematikou.

Jak se stane, že se informatik začne zajímat o biologii?
Biologie mě zajímala odjakživa, ale více jsem se k ní dostal až během doktorského studia softwarových systémů na Matfyzu. Pracoval jsem na vývoji metod pro rychlé vyhledávání v databázích proteinových sekvencí a struktur. Můj výzkum, který jsem dělal pod vedením Tomáše Skopala, dnešního prorektora pro informatiku na UK, byl ale hodně informaticky orientovaný. Tomáš Skopal se zabýval metrickým indexováním, což jsou speciální typy indexovacích struktur, a hledal aplikace těchto metod. Velké databáze biologických entit byly pro to ideálním objektem.
Tenkrát jsme vytvořili nástroj, který sice fungoval, ale k tomu, aby mohl být široce využíván v praxi, nebyl optimální. Hlavní problém byl v tom, že jsme ho vyvíjeli jako informatici bez většího biologického backgroundu. Biologické systémy mají různá omezení a pokud jim nerozumíte, může se stát, že vytvoříte něco, co na určitém typu dat funguje, ale na jiném už ne. Téma mě však zaujalo, takže jsem se mu dál věnoval a postupně do své expertizy zahrnul více i tu biologii.
Čím se tedy přesně bioinformatika zabývá?
Bioinformatika je obor, který kombinuje molekulární biologii a informatiku, konkrétně jde o aplikaci počítačových metod na molekulárně-biologická data, tj. na makromolekuly, jako jsou proteiny DNA a RNA. Studujeme jejich vzájemné interakce, ale také interakce s malými molekulami, což mohou být například léčiva.
Takže pomáháte vyvíjet nové léky?
Počítačový vývoj léčiv je jedna z často zmiňovaných aplikací bioinformatického výzkumu. Další velkou oblastí, kde se bioinformatický výzkum uplatňuje, je sekvenování DNA (sekvenací se zjišťuje pořadí nukleotidů v určitém úseku DNA; pozn. red.). Sekvenátory poskytují výzkumníkům raw data, která se musí roztřídit a ukládat, aby je bylo možné interpretovat a dále s nimi pracovat – to je dnes z velké části informatická úloha. V současnosti se většina dat z experimentů nahrává do veřejně přístupných databází, což jsou obrovské banky schraňující miliony biologických údajů. Tyto databáze jsou užitečné, protože umožňují porovnávat data z různých výzkumů napříč roky a objevovat souvislosti, které by jinak zůstaly skryté.
Bioinformatika je relativně nový obor, který se ale rychle rozvíjí. Kam se od doby, kdy jste se jí začal věnovat, posunula? V čem je dnes největší rozdíl?
Dnes máme k dispozici daleko více dat, než tomu bylo před 15 lety. Velký posun nastal v technologiích, zejména díky metodám strojového učení, které nám otevřely dveře a poskytly množství nových relativně kvalitních dat. Příkladem z poslední doby je nástroj AlphaFold na předpovídání 3D struktury proteinů. Loni za něj jeho tvůrci ze společnosti Google DeepMind dostali Nobelovu cenu za chemii.
V čem je tento nástroj tak přelomový, že si vysloužil nejvyšší vědecké ocenění?
Přelomový je v tom, že s jeho pomocí je teď možné vygenerovat věrohodnou proteinovou strukturu pro prakticky libovolnou sekvenci, což předtím nebylo možné. Tradiční experimentální metody pro zjišťování proteinové struktury, jako je například rentgenová krystalografie, jsou totiž značně časově i finančně náročné. Databáze PDB, která vznikla v 70. letech minulého století, v současnosti schraňuje jen nějakých 200 tisíc proteinových struktur získaných experimentálními metodami. To sice nezní jako úplně nízký počet, ale pro základní výzkum to může být omezující, navíc obsažená data jsou specifická a obsahují třeba ve velké míře proteiny asociované s různými lidskými nemocemi.
Naproti tomu stojí databáze UniProt, která obsahuje stovky milionů proteinových sekvencí. Sekvenátory jsou dnes vcelku běžným laboratorním vybavením, takže data o DNA a následně proteinové sekvenci jsou daleko dostupnější než údaje o proteinové struktuře. Cílem výzkumů proto dlouho bylo najít způsob, jak zásobu sekvenčních dat využít a vymyslet výpočetní metodu, která by dokázala proteinovou strukturu odvodit ze sekvence. To se právě podařilo s nástrojem AlphaFold.
Před 14 lety jste v rozhovoru pro univerzitní časopis zmínil, že „zlatým grálem“ studia proteinů je právě tohle – umět předpovídat ze sekvence proteinovou strukturu. Tento problém už tedy vědci vyřešili?
Pouze částečně, ukazuje se, že svět proteinů je daleko dynamičtější, než jsme si mysleli. Protein je sekvence aminokyselin, která se v 3D prostoru skládá do více méně rigidního tvaru. Poměrně velká část proteomu však obsahuje tzv. vnitřně neuspořádané regiony. To jsou části proteinů, kde se struktura stane stabilní až při nějakém vnějším impulzu, například navázání nějakého „partnera“, nějaké molekuly. Tohle však predikční metody nejsou schopny zachytit. Skutečnost, že proteiny jsou částečně nestrukturované, začaly výzkumy více reflektovat až v posledních pěti deseti letech. Ne že by se to dříve nevědělo, ale myslelo se, že je to v daleko menší míře. Dnes se předpokládá, že zhruba třetina lidského proteomu je nestrukturovaná. Proto teď vznikají další nové výpočetní metody pro predikci těchto nestrukturovaných částí.
Věda mi umožňuje dělat věci, které přede mnou ještě nikdo nezkoušel. To mě baví a naplňuje. A taky mi vyhovuje, že ve vědě mohu uspokojit svoji soutěživou povahu, třeba když se snažím vyvinout metodu, která bude úspěšnější než ty ostatní…
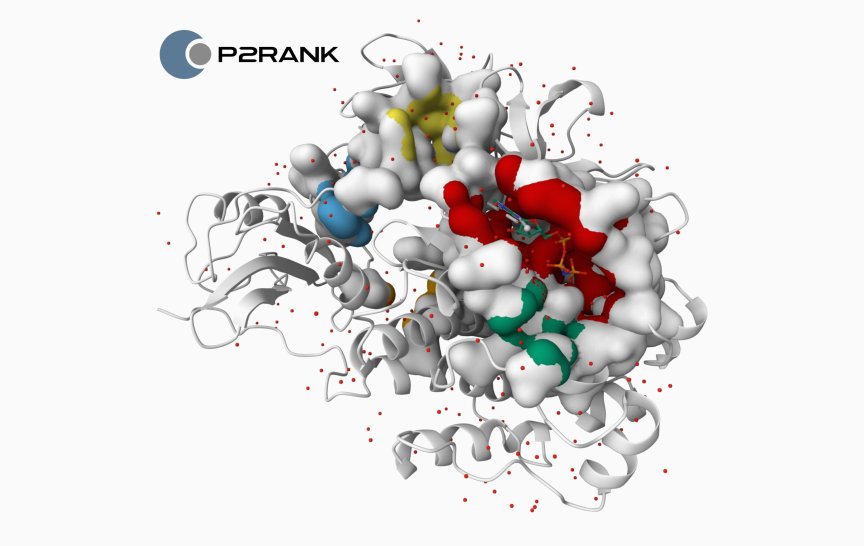
V minulosti jste pracoval na nástroji P2RANK, který využívá i známá evropská proteinová databáze PDBe (Protein Data Bank Europe). Čemu se věnujete teď?
Nástroj P2RANK slouží k detekci vazebných míst na povrchu proteinové struktury a jejich interakce s malými molekulami. Tento nástroj pořád udržujeme a vylepšujeme. Zároveň máme několik dalších odvozených projektů, nedávno jsme například získali grant na vývoj metody strojového učení pro odhalování kryptických (skrytých) vazebných míst.
Dalším projektem je portál G2P, pomocí kterého je, kromě jiného, možné vizualizovat najednou proteinovou sekvenci a strukturu a mapovat na tato data informace o variantách z genomové úrovně. Metoda využívá data z několika veřejně dostupných databází a propojuje informace z různých částí DNA a proteinové sekvence, které mapuje do 3D struktury. Jde o projekt, na kterém spolupracujeme s kolegy z Broad Institute, společného biomedicínského výzkumného centra MIT a Harvardu.
O metodě G2P vám v loňském roce vyšel společně s kolegy článek v prestižním vědeckém časopise Nature. Jak jste se k této mezinárodní spolupráci dostal?
Na metodě využívané v rámci G2P jsem začal pracovat během svého tříletého postdoktorského pobytu v Centru pro systémovou biomedicínu v Lucembursku. Tento nástroj zaujal kolegy z Broad Institute, kteří se specializují na výzkum genetických variant. V „Broadu“ mají databáze s informacemi o jednotlivých genetických variantách a o tom, jestli jsou patogenní, tedy způsobují onemocnění, anebo populační, tzn. že se vyskytují v populaci, ale nemají negativní efekt. Společně jsme pak vytvořili portál, který vlastně propojuje informace ze tří úrovní – úrovně DNA, proteinovou sekvenci a strukturu.
Co to znamená pro vědce, kteří portál využívají při svých výzkumech?
Znamená to pro ně nemalé ulehčení práce. Všechny zmíněné komponenty sice předtím byly k dispozici, ale nebyly propojené, takže například nebylo možné dělat rozsáhlé analýzy. S tímto nástrojem, který je přístupný přes programové rozhraní, lze vytvořit analýzu třeba pro všechny geny v lidském organismu, dívat se na jednotlivé varianty a sledovat, v jakých regionech proteinové struktury se objevují. To má největší potenciál právě pro vývoj léčiv cílených na konkrétní genové mutace a onemocnění.
V době, kdy jste začínal, tušil jste, jak velký potenciál bioinformatika má?
Musím se přiznat, že když jsem nastupoval na doktorské studium, tak jsem vůbec neplánoval, že u vědy zůstanu. Dělal jsem to čistě proto, že mě to zajímalo. Na konci mého doktorského studia už ale začínalo být k dispozici více dat a objevovaly se také nové aplikace, ať už v genomice nebo proteomice. Já jsem od počátku tíhnul spíše ke strukturní bioinformatice a to do jisté míry platí dodnes.
Čím si vás věda získala?
Tím, že nabízí poměrně velkou svobodu a že mi umožňuje dělat to, co přede mnou ještě nikdo nedělal. To mě baví a naplňuje. A taky mi vyhovuje, že ve vědě mohu uspokojit svoji soutěživou povahu, třeba když se snažím vyvinout metodu, která bude úspěšnější než ty ostatní…
Naznačoval jste, že jste se biologii musel doučit takzvaně za pochodu. Dnešní studenti už to mají jednodušší díky studijnímu programu bioinformatika, který jste před lety na UK založil společně s Marianem Novotným z Přírodovědecké fakulty…
Neřekl bych úplně, že to studenti mají jednodušší. Bioinformatiku jsme zakládali jako společný studijní program Matfyzu a Přírodovědecké fakulty. Naši posluchači vlastně studují dva obory – informatiku a molekulární biologii, přičemž z obou mají ten těžší základ. Kromě jiného musejí například absolvovat předměty, jako je matematická analýza, programování nebo algoritmizace – stejně jako studenti, kteří studují na Matfyzu „pouze“ informatiku. Má to však tu výhodu, že naši absolventi získávají v obou oblastech skutečně kvalitní základy, což jim otevírá brány do řady oblastí na pracovním trhu.
Kam například?
Často to jsou biotechnologické firmy, jako je MSD, jedna z pěti největších farmaceutických firem, která má v Praze své vývojové centrum. Řada studentů zůstává v akademii, což je případ i naší úplně první absolventky, která působila několik let na ETH a Harvardu a teď pracuje jako výzkumnice na katedře buněčné biologie Přírodovědecké fakulty. Díky tomu, že naši absolventi mají plnohodnotné informatické vzdělání, uplatní se bez problémů i jako vývojáři u softwarových firem.
Je toho spousta, co se v bioinformatice dá dělat. Na co se chcete v blízkém budoucnu zaměřit vy?
Hodně mě teď zajímá zmíněná dynamika proteinových struktur, což je věc, která výrazně mění náhled na data a na to, jak s nimi pracovat. Další téma, které má obrovskou perspektivu, je propojování strukturních dat na genomickou úroveň. Díky strojovému učení je teď možné analyzovat genomickou úroveň v kontextu znalostí o 3D proteinové struktuře, ale dá se jít ještě dál. V Lucembursku například vznikají projekty zaměřené na tzv. „disease maps“, což je něco jako Google mapy, akorát místo měst jsou na těchto mapách proteiny a malé molekuly. Tyto mapy zobrazují celý molekulárně-biologický systém, jednotlivé vrstvy, entity a jejich interakce, což umožňuje podívat se na problém v daleko širším kontextu. Například můžeme zjistit, co se stane v celém systému při vypnutí určitého genu. Integrace všech vrstev je zkrátka strašně zajímavá a díky pokrokům v technologiích se dá pronikat hlouběji.
Když vás tak poslouchám, říkám si, že vám asi pracovní vyhoření nehrozí…
Doufám, že ne, a pokud ano, tak nejspíš dělám něco špatně.
doc. RNDr. David Hoksza,
Ph.D.
Vystudoval informatiku (obor softwarové systémy) na MFF UK. V letech
2017–2020 působil jako postdoktorand v Luxembourg Centre for Systems
Biomedicine, University of Luxembourg. Od roku 2021 je docentem na katedře
softwarového inženýrství MFF UK. Zabývá se výzkumem v oblasti strukturní
bioinformatiky a vizualizací dat. Mezi jeho hlavní současné projekty patří
framework P2Rank, R2DT a portál Genomics 2 Proteins. Na Univerzitě Karlově
spoluzaložil bakalářský a magisterský studijní program Bioinformatika, je
také garantem nového doktorského programu Bioinformatika a výpočetní
biologie.
Mohlo by vás také zajímat:
Václav
Rozhoň: Věda je sociální proces
Matematik
Dalimil Peša: Vždycky mě bavilo přicházet věcem na kloub
Filip
Dvořák: Lidská mysl není pro AI nejlepším vzorem, v budoucnu objevíme
efektivnější architektury